






AntPhos: 为高效偶联及不对称偶联打造的单膦配体


AntPhos: 为高效偶联及不对称偶联打造的单膦配体

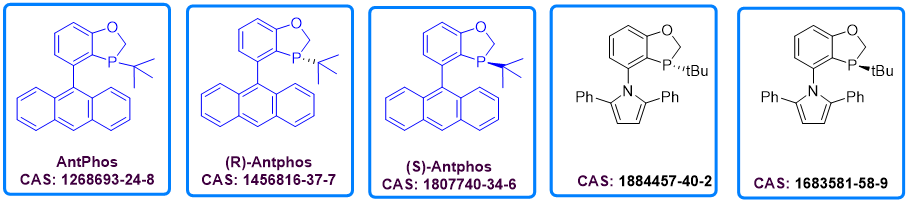
在P-手性二氢苯并氧磷杂环戊烷的C-4位引入蒽基即得到AntPhos。自其被发现以来,AntPhos已经广泛应用在偶联、硼化、环化等多种过渡金属催化反应中。AntPhos具有独特而显著的结构特征和理化性质:1) AntPhos的苯并磷氧杂环具有稳定而明确的构象; 2) AntPhos的蒽基部分具有与过渡金属潜在配位的能力,从而使AntPhos 实现低催化当量情况下的偶联反应; 3) AntPhos的相对富电子结构使能有效活化惰性化学键; 4) AntPhos的大π体系能与底物产生非共价作用,从而使手性AntPhos在很多不对称催化中有表现出高立体选择性; 5) AntPhos对空气稳定,因此非常便于在工业生产中使用。
1.Miyaura 硼化[1]
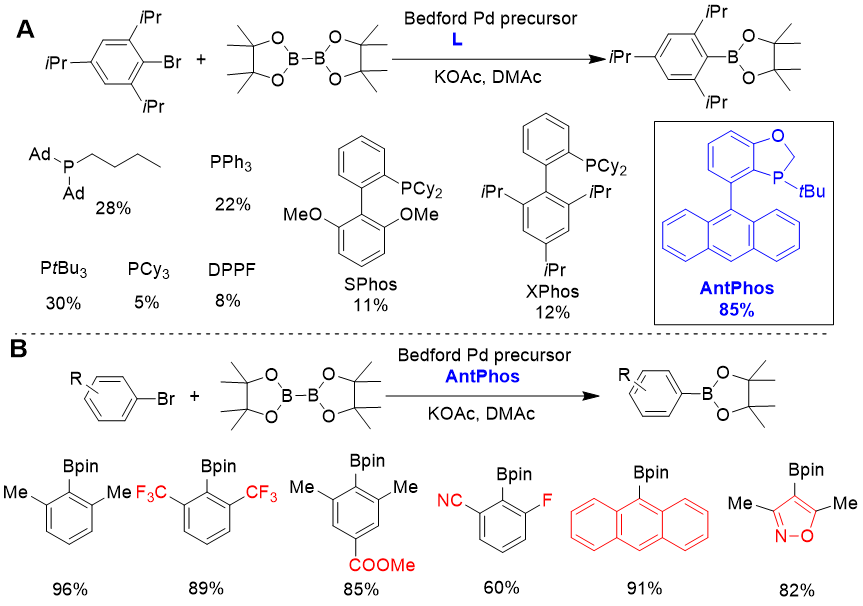
图1. Pd-AntPhos 催化的Miyaura 硼化
由于Suzuki-Miyaura反应在构建碳碳键方面的重要地位,芳基硼化物成为了关键
的合成砌块,在药物合成领域具有重要的应用价值。Pd-AntPhos体系能催化大位
阻芳基溴的Miyaura-硼化。如图1A所示,由于底物位阻大,大量著名的常用配体
均只能实现低产率的硼化,而AntPhos则显示了在大位阻底物Miyaura-硼化反应
中的优越性,能以高产率获得相应芳基硼酸酯。该方法对于含三氟甲基、氰基、酯
基等重要官能团的底物,或者含并环、杂环的底物均能兼容,显示出良好的底物普
适性(图1B),因此适合复杂分子或药物分子在合成后期使用。
2.大位阻Suzuki-Miyaura偶联[2,3]
AntPhos非常适用于大位阻芳基-芳基偶联。底物中含有硝基、氰基、醛基
等重要且敏感的官能团均能兼容,而且·并环或杂环不受影响[2,3]。该方法的
实用性在天然产物parnafungin A1的全合成中作为关键的一步得到论证。
AntPhos也能催化一个复杂的邻位具有游离酚羟基的芳基硼酸半酯与一个含
硝基的溴苯偶联(图2B)[3]。
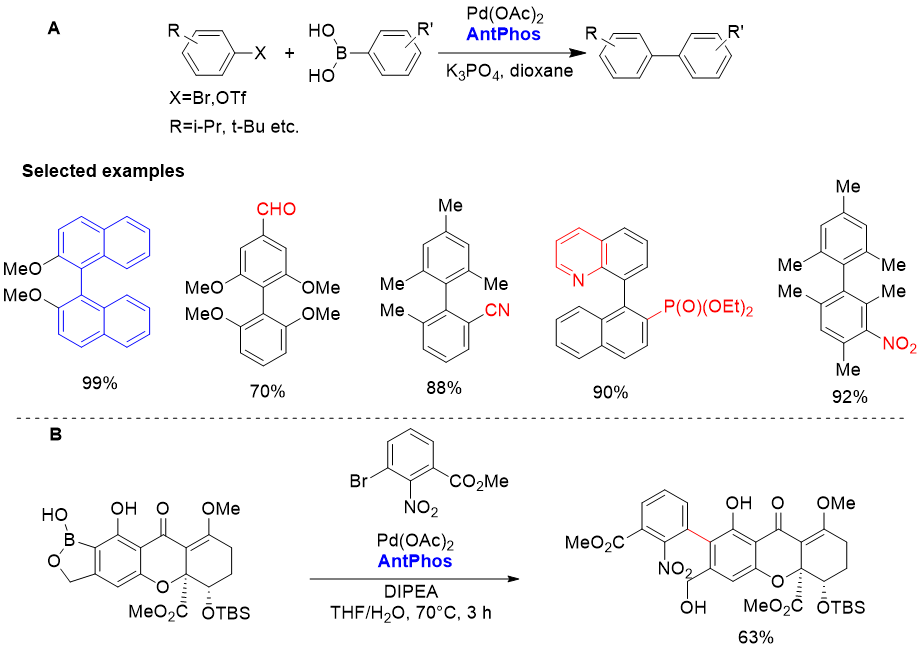
图2. Pd-AntPhos 催化的大位阻Suzuki-Miyaura交叉偶联
3. 芳基-烷基Suzuki-Miyaura交叉偶联 [4]
芳基-烷基Suzuki-Miyaura偶联是一类难度大却很重要的偶联反应,尤其是
邻位双取代芳基卤化物与烷基硼酸(或硼酸酯)的偶联挑战巨大。需要解决的
问题包括:1)该偶联反应需要克服两个偶联片间较大的位阻,;2)由于烷基
硼酸存在β-氢,该偶联反应需要有效抑制容易发生的β-氢消除。AntPhos成
功克服了上述难点,为大位阻芳基-烷基偶联提供了有效的方法。AntPhos
能实现邻位二取代芳基卤化物与一系列环状二级硼酸化合物的偶联,相应的三
元环、四元环、五元环、六元环硼酸类底物均可采用该方法实现偶联,该方法
强大的偶联能力在合成两种齿轮状分子中得到充分的体现。

图3.大位阻邻位二取代芳基卤化物与烷基二级硼酸的高效Suzuki-Miyaura偶联
4.低催化当量的Suzuki-Miyaura 偶联[5]

图4. 低催化当量的Suzuki-Miyaura 偶联
在此偶联案例中,老工艺采用Pd-PPh3催化体系,催化剂当量需要1 mol %,
且后处理需要三次重结晶才能获得符合纯度要求的产品。而Pd-AntPhos 催化体
系实现了40,000的催化转化数(TONs),即33g钯和55g配体实现1.5吨产品的
制备,且后处理仅需一次结晶获得高纯度产品。与老工艺相比,新工艺极大缩减了
生产成本,实现工艺升级。

1. 炔酮的不对称环化反应[6,7]

图5. Ni-AntPhos催化的炔酮不对称还原环化反应
炔酮类底物在Ni-AntPhos的催化下能发生还原环化反应,获得一系列含叔醇
的手性四氢呋喃、四氢吡咯、四氢吡喃类化合物。该反应产率高,对映选择性
好,底物普适性广,适合多种具有高附加值的杂环化合物合成。[6,7]
2. 不对称去芳构环化反应 [8-10]
近年来,去芳构环化反应已得到大量的研究,并应用在多种天然产物的全合成
中。研究人员发现,以AntPhos或其同类单膦配体与钯组成的催化体系能实现
分子内的高效不对称去芳构环化反应,构建6/n/6并环骨架(n=5, 6, 7)。
该化学反应具有高收率、高化学选择性、高对映选择性及底物普适性广的优点,
并成功应用于多种萜类、甾体类和生物碱类天然产物的合成中。如图6A所示,
在最初的条件筛选中,几种P-手性二氢苯并氧磷杂环戊烷磷配体均以较好的产
率和对映选择性实现了分子内的去芳构环化反应,得到相应的产物,但最终以
2,5-二苯基-吡咯取代的配体效果最佳,能以94%的产率,96:4的区域选择性
及92%的ee获得目标产物[8]。进一步研究发现,该方法在多类手性多环化合物
的合成中表现出极高的效率,展示出重要的应用价值,。研究团队采用该方法
以简短的路线先后完成了生物碱类天然产物(-)-crinine,
(-)-aspidospermidine克级规模的合成,(-)-minfiesine的形式合成, 以
及免疫抑制剂(+)-dalesconol A 和 B的简洁全合成。在这些研究中,不管是
反应活性或是对映选择性,AntPhos都扮演了极为重要的角色(如图6B所示)
[9-10]。

图6. Pd-AntPhos催化不对称去芳构环化反应
3. 不对称Heck 反应[11-15]

图7. Pd-AntPhos 催化的不对称 γ-芳基化反应
手性AntPhos可用于不对称Heck反应,构建手性季碳中心,并在天然产物
Mesembrine的不对称全合成中得到了应用[11]。复旦大学的研究团队则用类
似的催化体系,通过Heck反应完成了一系列的γ-丁烯内酯和γ-丁内酯的构建,
该反应条件温和,化学选择性、对映选择性、反应产率及底物普适性都很理想
[12]。

图8. Pd-AntPhos catalyzed cascade cross-coupling reactions13-15
1,1-双取代烯酰胺底物在Pd-AntPhos的催化下,可以实现烯基的双官能团化
合成异吲哚啉-1-酮类化合物,推测该反应机理可能是烯酰胺与芳基(或烯基)
硼酸经历了串联Heck/Suzuki偶联反应(如图8A所示)[13]。同样以Pd-
AntPhos为催化体系,具有烯酰胺取代的联苯类底物可通过串联Heck/羰基化
/胺基化反应高对映选择性的构建大量具有轴手性的连芳基[b,d]氮杂䓬类化合
物(如图8B所示)[14-15] 。
4. 不对称烯烃的芳基/芳氧基化反应[16]

在Pd-AntPhos体系的催化下,烯基可实现芳基/芳氧基双官能团化反应,构
建大量含有手性季碳中心的1,4-苯并二噁烷、1,4-苯并二噁嗪及苯并吡喃类化
合物,对映选择性及收率均较为优异。
AntPhos已广泛应用于Suzuki-Miyaura偶联反应,尤其是在大位阻底物
的偶联反应中具有独特的优势,具有底物普适性好、官能团容忍度高、催化剂
载量低、适合工业化放大等优点。手性AntPhos则在多类不对称催化反应中得
到了应用,表现出优秀的活性和对映选择性。由于具有既高效又用途广泛的特
点,AntPhos在科学研究及工业生产中均已得到广泛的应用,是医药、农药及
新材料研发中不可缺少的高效膦配体!目前绍兴赜军生物医药科技有限公司已
实现消旋及两种构型AntPhos年数十公斤级的供应能力,产品面向全球。
[1] Org. Lett. 2011, 1
[2] Chem. Eur. J. 2013, 19, 2261-2265
[3] Chem. Sci. 2021, 12, 10313-10320.
[4] Org. Chem. Front. 2014, 1, 225-229
[5] An example in a metric ton scale.
[6] Angew. Chem., Int. Ed. 2015, 54, 2520-2524
[7] J. Org. Chem. 2021, 86, 5166-5182.
[8] Angew. Chem., Int. Ed. 2015, 54, 3033-3037
[9] Chem. Sci. 2017, 8, 6247-6256.
[10] J. Am. Chem. Soc. 2017, 139, 3360-3363.
[11] J. Org. Chem. 2016, 81, 10165-10171
[12] Angew. Chem., Int. Ed. 2022, 61, e202202046.
[13]Org. Chem. Front. 2021, 8, 4106-4111
[14] Org. Lett. 2021, 23, 3636-3640.
[15] Org. Chem. Front. 2022, 9, 939-945.
[16] Angew. Chem., Int. Ed. 2016, 55, 5044-5048
*Kindly remind: more products information please visit www.zejunpharma.com



